tyRa Vignette
Daniel Sprockett
tyRa.RmdtyRa is an R package that I created to facilitate building models from 16S rRNA amplicon data stored in phyloseq objects.
Import
Import the phyloseq object. We’ll just take the samples from healthy individuals for this demonstration. We’ll also filter down to only very prevalent taxa.
ps <- readRDS("/Users/daniel/R_packages/tyRa/data/infants_ps.rds")
ps
#> phyloseq-class experiment-level object
#> otu_table() OTU Table: [ 1405 taxa and 200 samples ]
#> sample_data() Sample Data: [ 200 samples by 207 sample variables ]
#> tax_table() Taxonomy Table: [ 1405 taxa by 8 taxonomic ranks ]
#> phy_tree() Phylogenetic Tree: [ 1405 tips and 1404 internal nodes ]
#> refseq() DNAStringSet: [ 1405 reference sequences ]This dataset is a subset of gut microbiome profiles generated from European infants. It had already been rarefied to 10,000 sequences per sample.
Fit Model
Currently, there is only model available, but I’ll be adding more over time. We’ll fit this dataset with the Sloan (2006) Neutral Community Model from Burns et al. (2016).
spp.out <- tyRa::fit_sncm(spp = otu_table(ps)@.Data, pool=NULL, taxon=data.frame(tax_table(ps)))
#> Waiting for profiling to be done...Plot
Now we’ll plot the output.
plot_sncm_fit(spp.out, fill = NULL, title = "Model Fit")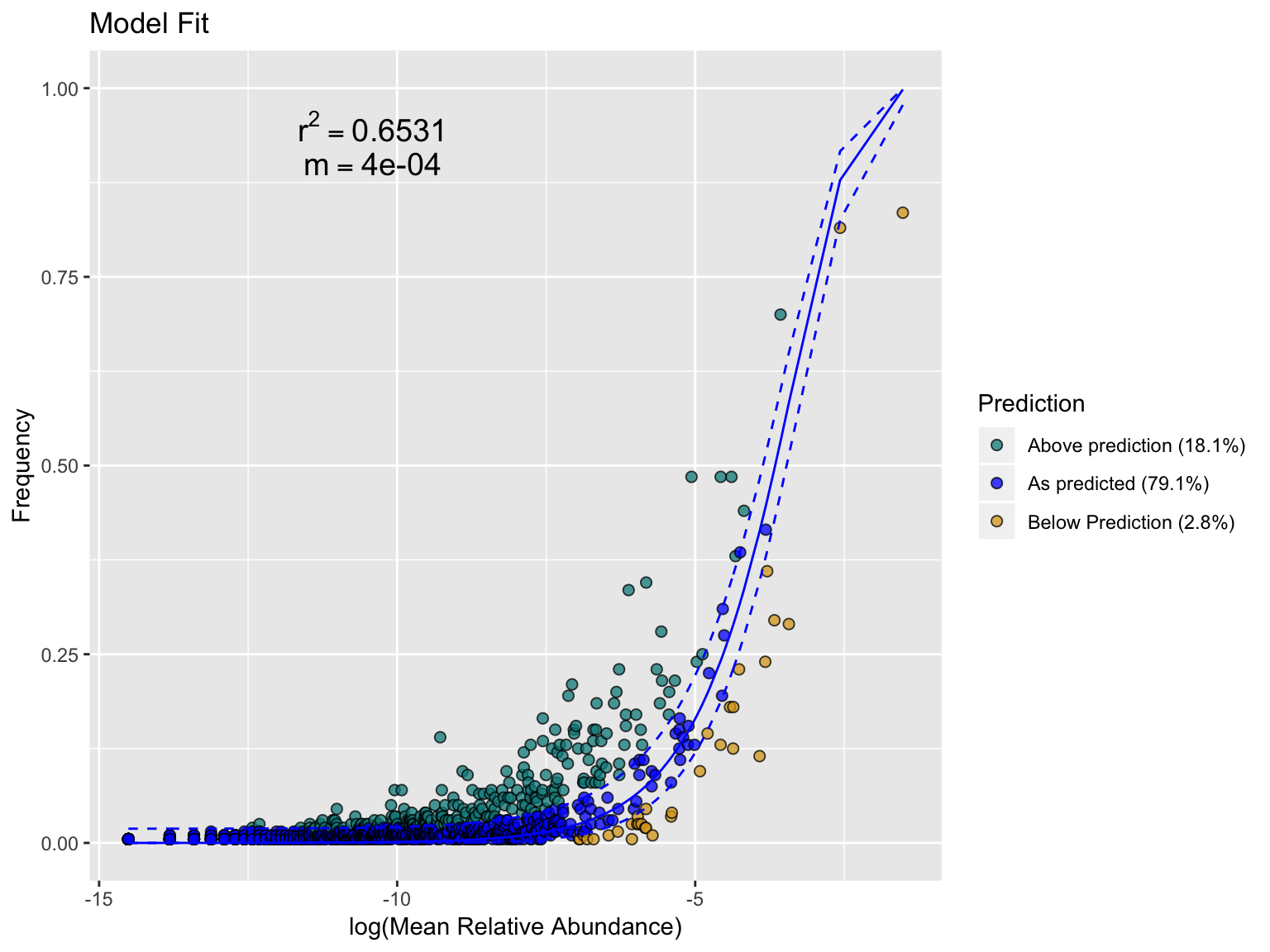
As you can see from this plot, nearly 80% of the ASVs in this dataset fit the neutral model.
This is a work in progress, so please let me know if you run into any problems. Thank you!